Beta thalassemia
Beta thal – variable levels of beta chain synthesis; 2 genes on chromosome 11
Presentation depends on type of mutation
β – Normal production
β+ – Decreased β chain production
β0 – No β chain production
Damage is from precipitation of α chains
β/β+ and β/β0
“Silent carrier or beta thal minor”
Mild anemia (2-3 g/dL below normal)
Low MCV
Often misdiagnosed as Fe deficiency
Mentzer index (MCV/RBC #) <13
Increased Hgb A2 (α2δ2) on Hgb
electrophoresis
β+/β+
“Thalassemia intermedia”
Moderate anemia
(Hgb 6-8 mg/dL)
Moderate elevation of Hgb F (60-80%)
β Thalassemia Major (β0/β0) aka Cooley anemia
Present in 2nd 6mos of life
Severe hemolytic anemia
Extramedullary hematopoesis
HSM
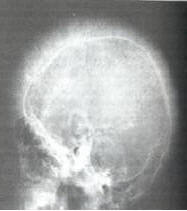
Bone marrow, face/skull bones expand
Green/brown skin color
Pallor
Jaundice
Hemosiderosis
Pancreas (DM)
Heart (arrhythmia, CHF)
Laboratory findings
Severe hypochromic, microcytic anemia
Poikilocytosis, target cells
Hgb <5
Unconjugated bilirubin elevation
TIBC low (all sites bound)
Fe level high
Fetal hemoglobin level: >90%
Therapy
Regular transfusions (q 4-5 weeks)
Deferoxamine (Fe chelator)
Splenectomy
Indicated for hypersplenism or progressive
enlargement
BMT
Picture: frontal bossing, maxillary hyperplasia, (2) Hair on end skull